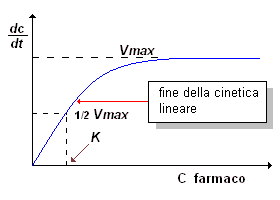
 I modelli farmacocinetici finora discussi sono fondati sull'ipotesi che le velocità con cui i farmaci entrano, si distribuiscono e vengono eliminati dall'organismo seguano una cinetica di ordine primo, cioè siano funzioni lineari delle concentrazioni di principio attivo. Questo significa che le concentrazioni plasmatiche dei farmaci sono considerate proporzionali alla dose: quando la proporzionalità non è più rispettata, la cinetica cessa di essere lineare e in questa condizione si applica il modello farmacocinetico di Michaelis-Menten, il cui andamento è riportato nel grafico a destra.
I modelli farmacocinetici finora discussi sono fondati sull'ipotesi che le velocità con cui i farmaci entrano, si distribuiscono e vengono eliminati dall'organismo seguano una cinetica di ordine primo, cioè siano funzioni lineari delle concentrazioni di principio attivo. Questo significa che le concentrazioni plasmatiche dei farmaci sono considerate proporzionali alla dose: quando la proporzionalità non è più rispettata, la cinetica cessa di essere lineare e in questa condizione si applica il modello farmacocinetico di Michaelis-Menten, il cui andamento è riportato nel grafico a destra.
Il passaggio ad una cinetica non lineare è conseguente al fatto che il farmaco, distribuendosi nell'organismo, può incontrare stadi saturabili, ove la velocità non aumenta più con la dose. Queste situazioni si verificano essenzialmente in quattro casi:
In questi casi, lo studio cinetico dell'andamento concentrazione-tempo del farmaco nel plasma risulta matematicamente complesso; tuttavia, il passaggio ad una cinetica non lineare, dà ragione di alcune menifestazioni tossicologiche che possono presentarsi:

Nel caso dei medicinali equivalenti, la cinetica non lineare ed un modesto indice terapeutico dovrebbero essere valutate attentamente in quanto, come discusso, la biodisponibilità fra originator e medicinale equivalente è compresa in un range di accettabilità.
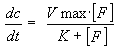 (eq. 1) (eq. 1) |
dove:
dc/dt = velocità di trasporto del farmaco;
Vmax = velocità massima raggiunta con la saturazione del carrier;
K = costante di affinità tra farmaco e carrier;
L'equazione di Michaelis-Menten è stata utilizzata per descrivere la saturazione nel trasporto del farmaco attraverso la membrana; la stessa equazione, con il segno negativo, può essere utilizzata per descrivere la diminuzione dei livelli plasmatici del farmaco.
A concentrazioni di farmaco elevate (F » K), la (1) si riduce ad una equazione differenziale di ordine zero secondo cui la concentrazione plasmatica diminuisce con velocità costante pari a Vmax = Km (la massima velocità metabolica)
dc/dt = dF/dt = - Vmax = - Km che integrata fornisce F = F0 - Km · t (eq. 2)
A concentrazioni molto basse (F « K), la concentrazione plasmatica diminuisce con un andamento descritto da una equazione differenziale di primo ordine:
dc/dt = dF/dt = - F(Vmax)/(K) = - F · Km che integrata fornisce F = F0 e-Km t (eq. 3)
E' stato ipotizzato che tutti i farmaci metabolizzati a livello epatico siano soggetti alla stessa farmacocinetica enzimatica secondo Michaelis-Menten. Questo significa che a determinate alte concentrazioni tutti i farmaci dovrebbero presentare una cinetica non lineare. D'altra parte, l'intervallo terapeutico delle maggior parte dei farmaci ricade nella parte inferiore della curva di M-M e quindi segue una cinetica descritta dall'eq. 3: questo spiega perché la loro concentrazione plasmatica in funzione del tempo ha andamento esponenziale.
Per esempio, la teofillina ha mostrato una cinetica non lineare a concentrazioni elevate, ma lineare a concentrazioni più basse.
 Un tipico esempio di cinetica non lineare è offerto dall'alcol etilico. Il 90-95% della quantità di alcol assunta (dall'uomo) viene metabolizzata a livello epatico, la rimanente parte a livello del tratto digerente (in particolare lo stomaco), del rene, dei polmoni e dei muscoli. L'alcol assunto in dosi elevate (un adulto di 70 kg circa può consumare in più riprese circa un litro di vino a 12° nell'arco di 24 h) viene eliminato con una cinetica di ordine zero (quindi a velocità costante, indipendentemente dalla quantità di alcol presente) in quanto i sistemi enzimatici deputati all'ossidazione di etanolo in acetaldeide vengono rapidamente saturati.
Un tipico esempio di cinetica non lineare è offerto dall'alcol etilico. Il 90-95% della quantità di alcol assunta (dall'uomo) viene metabolizzata a livello epatico, la rimanente parte a livello del tratto digerente (in particolare lo stomaco), del rene, dei polmoni e dei muscoli. L'alcol assunto in dosi elevate (un adulto di 70 kg circa può consumare in più riprese circa un litro di vino a 12° nell'arco di 24 h) viene eliminato con una cinetica di ordine zero (quindi a velocità costante, indipendentemente dalla quantità di alcol presente) in quanto i sistemi enzimatici deputati all'ossidazione di etanolo in acetaldeide vengono rapidamente saturati.
Un altro esempio*, più complesso, è utile per comprendere l'importanza clinica della cinetica non lineare. Ad un bimbo di 6 anni in trattamento con fenitoina (antiepilettico), viene associata la somministrazione di acido valproico per un controllo migliore degli attacchi. In séguito all'insorgenza di segni di tossicità alla fenitoina (atassia) si procede con urgenza ad una determinazione della fenitoina che fornisce il valore di 15,0 mg/L (intervallo terapeutico 10-20 mg/L). Anche il valore al picco prelevato 5 ore dopo l’ultima dose è 18,0 mg/L (accettabile), e pure l’acido valproico misurato su entrambi i campioni è nel range terapeutico. Il controllo di qualità del farmaco rientra nei limiti di accettabilità.
L'effetto tossico associato ad una concentrazione plasmatica di fenitoina pur entro i limiti terapeutici (l’atassia si manifesta solitamente ad alte concentrazioni di fenitoina plasmatica, generalmente intorno a 30-40 ug/mL) può sembrare anomalo tanto da far pensare a una reazione avversa individuale o a probabili errori analitici.
La concentrazione plasmatica di fenitoina è un classico farmaco secondo M-M e vari schemi di dosaggio sono stati basati su questo concetto. Così, la spiegazione del quadro clinico in questione, potrebbe individuarsi nel fatto che la fenitoina è caratterizzata da una cinetica di saturazione, in base alla quale l'esaurimento del sistema enzimatico preposto al metabolismo del farmaco provoca accumulo della forma farmacologicamente attiva (fenitoina libera) con conseguente comparsa di fenomeni tossici. Tuttavia, nel caso proposto, la concentrazione plasmatica del farmaco al picco (18 mg/L) non è particolarmente elevata, per cui la diminuzione della clearance (velocità di eliminazione) non è probabilmente la causa della tossicità. E allora?
Allora, la causa va piuttosto cercata nella possibile diminuzione dei siti di legame con l'albumina impegnati anche dall’acido valproico che, competendo con la fenitoina fa sì che la concentrazione di picco - accettabile in condizioni di non competizione - diventi pericolosa. Questo significa che la concentrazione plasmatica al picco (18 mg/L) fornisce un'indicazione fuorviante di quella che è la reale concentrazione attiva. Infatti, la fenitoina è legata per circa il 90% alle proteine plasmatiche, per cui la frazione libera - farmacologicamente attiva - è molto ridotta (circa 10%), tanto che modesti cambiamenti nel legame con le proteine plasmatiche possono provocare un grande cambiamento nella quota libera del farmaco. Così, per esempio, un 5 % di riduzione del legame con le proteine plasmatiche dovuta alla presenza di acido valproico, provoca un aumento dei livelli di fenitoina libera del 50% (la frazione libera passa dal 10 al 15%).
Riassumendo: l'associazione dei due farmaci antiepilettici in questione (fenitoina e acido valproico) comporta lo spiazzamento della fenitoina dall'albumina ad opera dell'acido valproico, con conseguente aumento di concentrazione di fenitoina libera. L'incremento della frazione di fenitoina libera determina una più elevata clearance epatica del farmaco e quindi una riduzione della sua concentrazione plasmatica totale. A séguito dell’esaurimento delle riserve epatiche (saturazione indotta dall'acido valpronico) deputate al metabolismo della fenitoina la sua frazione libera tende ad aumentare e, poiché questa rappresenta la forma farmacologicamente attiva del farmaco, il suo accumulo dà origine all’effetto tossico osservato (atassia). In questo caso particolare, la tossicità del farmaco è dovuta all’aumento della percentuale della frazione libera piuttosto che ad un sovradosaggio del farmaco stesso. Le determinazioni della concentrazione plasmatica della fenitoina confermano questo fatto in quanto i dosaggi eseguiti, compreso quello di picco, rientrano nel range terapeutico pur constatando tossicità.
A questo punto per controllare il trattamento terapeutico sarebbe di determinante aiuto eseguire il monitoraggio della concentrazione ematica di fenitoina libera (difficilmente attuabile nell’attività routinaria di laboratorio). In ogni caso la sospensione della somministrazione dell’acido valproico e il controllo della concentrazione di fenitoina ematica dovrebbe portare alla scomparsa dell’effetto tossico indesiderato.
(*) P. Bonvicini, A. Croce, B. Forcina - Casi Clinici e Quesiti di Medicina di Laboratorio, 2004
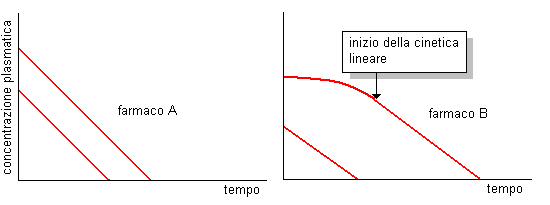
Nelle figure sopra, sono riporati i profili farmacocinetici riferiti a due farmaci A e B, il secondo dei quali ad elevate concentrazioni segue una cinetica non lineare. Riportando il logaritmo delle concentrazioni plasmatiche, Cp, in funzione del tempo per il farmaco A (somministrato a due dosaggi differenti) si ottengono due rette parallele, in accordo col fatto che la velocità di eliminazione è proporzionale alla dose; per il farmaco B (somministrato a due dosaggi differenti), si ottengono due rette parallele solo dopo che il farmaco somministrato a dosaggio maggiore segue una cinetica lineare.
| 10 | ||||||||||
Marcello Guidotti, copyright 2007-2011
questa pagina può essere riprodotta su qualsiasi supporto o rivista purché sia citata la fonte e l'indirizzo di questo sito (ai sensi degli artt. 2575 e 2576 cc. Legislazione sul diritto d'autore). Le fotografie sono tratte da siti web e sono, o possono ritenersi, di pubblico dominio purché utilizzate senza fini di lucro. Le immagini di prodotti presenti nel sito hanno unicamente valenza esemplificativa oltre che, eventualmente, illustrare messaggi fuorvianti e non vi è alcun richiamo diretto o indiretto alla loro qualità e/o efficacia il cui controllo è affidato alle autorità regolamentatorie.