| Indicazione | volume approssimativo di solvente in ml per g di sostanza |
| solubilissimo | meno di 1 |
| molto solubile | da 1 a 10 |
| solubile | da 10 a 30 |
| moderatamente solubile | da 30 a 100 |
| poco solubile | da 100 a 1000 |
| molto poco solubile | da 1000 a 10000 |
| praticamente insolubile | più di 10000 |
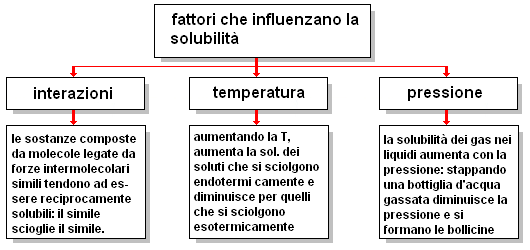
Come si vede, la solubilità delle sostanze varia ampiamente e, ad eccezione delle sostanze solubilissime in un adatto solvente, la solubilità varia all'interno della stessa classe: per es., 1 g di una sostanza molto solubile può disciogliersi in un volume che varia da 1 a 10 ml.
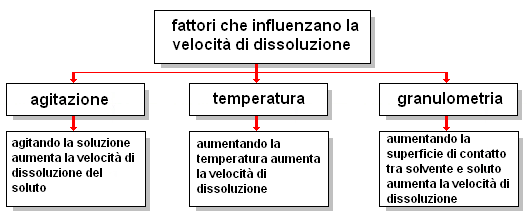
Un altro elemento che deve essere considerato è che le classi di solubilità riportate in tabella non fanno riferimento al tempo necessario per la dissoluzione. Dunque, per definire il concetto di solubilità occorre:
Oltre ai fattori riassunti negli schemi precedenti, occorre considerare che i sali presentano valori di solubilità diversi a seconda delle combinazioni catione - anione. Il grafico che segue mostra la variazione di solubilità con la temperatura.
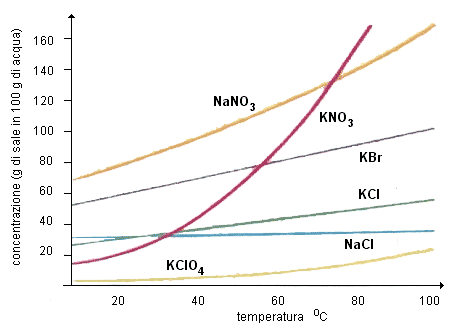
Dal grafico risulta evidente la differente solubilità di KCl e KBr. Questo differente comportamento è correlabile alle dimensioni degli ioni ed alla solvatazione con le molecole del solvente.
 |
 |
 |
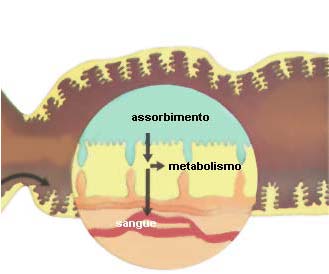
La figura sopra, illustra i processi che possono far variare l'assorbimento di un farmaco nell'intestino: tempo di svuotamento gastrico e di transito intestinale; acidità del contenuto intestinale, area della superficie di assorbimento, stato dell'epitelio intestinale, caratteristiche del flusso sanguigno.
In particolare, i modelli matematici impiegati per descrivere il processo della dissoluzione (inverso della cristallizzazione), sono basati sull'equazione proposta nel 1897 da Noyes e Whitney:
| (1) |
dove:
q = quantità di solido liberata nel tempo t;
k = coefficiente di proporzionalità;
SS = superficie specifica delle particelle dissolte;
CS = concentrazione massima di saturazione (solubilità del principio attivo);
C = concentrazione del farmaco nel fluido biologico (compresa tra 0 e CS)
dall'equazione 1, risulta che, pur rimanendo costante la solubilità di una forma farmaceutica solida, la velocità di diffusione può essere variata sensibilmente aumentando la suddivisione del solido in particelle più piccole. In particolare, se le dimensioni delle particelle hanno l'ordine di grandezza di micrometri, si verifica anche un aumento di solubilità (legato all'aumento di interazioni solido-liquido).
Nernst e Brunner, nel 1904, esplicitando il coefficiente k della (eq. 1), hanno definito il concetto di "strato diffusionale" (figura sopra - circoletto rosso sulla compressa).
| (2) |
come si vede, l'eq. 2, connette la velocità di dissoluzione al gradiente di concentrazione.
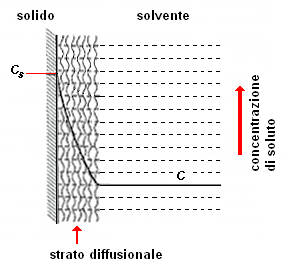 Il grafico a destra mostra l'andamento della velocità di dissoluzione attraverso lo strato diffusionale. Si può notare come in prossimità della forma solida, la concentrazione è quella di saturazione, CS, che decresce via via che raggiunge il confine esterno dello strato diffusionale, dove la concentrazione, C, si mantiene costante nell'ambiente gastrointestinale.
Il grafico a destra mostra l'andamento della velocità di dissoluzione attraverso lo strato diffusionale. Si può notare come in prossimità della forma solida, la concentrazione è quella di saturazione, CS, che decresce via via che raggiunge il confine esterno dello strato diffusionale, dove la concentrazione, C, si mantiene costante nell'ambiente gastrointestinale.
Lo strato diffusionale, è uno strato dinamico: continuamente le molecole che lasciano questo strato per entrare nei fluidi biologici, vengono rimpiazzate dalle molecole che abbandonano la compressa che si disaggrega. E' quindi evidente come l'aumento del gradiente di concentrazione, (CS - C)/h, ottenuto con un aumento della solubilità del farmaco, oppure con la riduzione dello strato diffusionale, favorisca il processo di dissoluzione.
Se si ammette che il farmaco viene prontamente assorbito appena entra nei fluidi biologici, la concentrazione C diviene trascurabile rispetto a CS e quindi l'eq. 2 assume la forma:
| (3) |
 Anche la granulometria delle polveri influenza l'azione dei farmaci. La griseofulvina è pochissimo solubile in acqua (1 mg/ml), ma riducendo di 3,8 volte le dimensioni delle sue particelle (polveri), si possono ottenere livelli ematici 2,3 volte superiori e dunque è possibile somministrare dosaggi minori. Ancóra, la sulfadiazina può essere somministrata come sospensione orale o nella forma micronizzata che permette un assorbimento superiore del 20%.
Anche la granulometria delle polveri influenza l'azione dei farmaci. La griseofulvina è pochissimo solubile in acqua (1 mg/ml), ma riducendo di 3,8 volte le dimensioni delle sue particelle (polveri), si possono ottenere livelli ematici 2,3 volte superiori e dunque è possibile somministrare dosaggi minori. Ancóra, la sulfadiazina può essere somministrata come sospensione orale o nella forma micronizzata che permette un assorbimento superiore del 20%.
![]()
al quale corrisponde l'equazione:
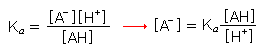
considerando che la concentrazione di saturazione è legata alla presenza del farmaco per le frazioni non ionizzata e ionizzata, si può scrivere: Cs = [AH] + [A-]
inserendo in questa equazione il valore di [A-] ricavato nell'equazione precedente, si ottiene:
![]()
poiché la concentrazione iniziale dell'acido è: Ci = [AH]
inserendo queste condizione nell'equazione precedente, si ricava la concentrazione di saturazione:
![]()
sostituendo nella (eq. 3), si ottiene:
(4) 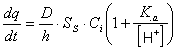 |
l'equazione così ottenuta mostra come la velocità di dissoluzione sia legata all'acidità dell'ambiente. Per questo, ove possibile, si possono migliorare le caratteristiche di una formulazione farmaceutica salificando il principio attivo. In questo caso Cs = [A-], cioé la velocità di dissoluzione dipende direttamente dalla concentrazione del farmaco (cfr. eq. 3).
La salificazione è il mezzo che consente di cambiare le proprietà chimiche e biologiche di un farmaco senza variarne la struttura; in questo modo è possibile migliorarne la cinetica, l'assorbimento e le proprietà chimico-fisiche (stabilità, igroscopicità, dissoluzione). Tuttavia, è bene considerare che sali diversi dello stesso principio attivo sono prodotti distinti, caratterizzati da un proprio profilo chimico e biologico differente i cui effetti non si possono prevedere con certezza e quindi non è corretto ritenere che due sali diversi dello stesso principio attivo si comportino esattamente allo stesso modo.
Altri fattori che possono influenzare la velocità di dissoluzione sono il polimorfismo e la presenza di tensioattivi; questi ultimi aumentano la velocità di dissoluzione in quanto favoriscono la bagnabilità delle polveri e ne aumentano la solubilità.
| 2 | |||
Marcello Guidotti, copyright 2003-2004-2009
questa pagina può essere riprodotta su qualsiasi supporto o rivista purché sia citata la fonte e l'indirizzo di questo sito (ai sensi degli artt. 2575 e 2576 cc. Legislazione sul diritto d'autore). Le fotografie sono tratte da siti web e sono, o possono ritenersi, di pubblico dominio purché utilizzate senza fini di lucro. Le immagini di prodotti presenti nel sito hanno unicamente valenza esemplificativa oltre che, eventualmente, illustrare messaggi fuorvianti e non vi è alcun richiamo diretto o indiretto alla loro qualità e/o efficacia il cui controllo è affidato alle autorità regolamentatorie.