fattori determinanti le modificazioni nella farmacocinetica
Come tutti i modelli, quelli descritti per le applicazioni farmacocinetiche sono per definizione riferiti a sistemi ideali: riflettono le semplificazioni fatte per formulare le equazioni di volta in volta proposte. Sebbene i sistemi ideali si discostino dalla realtà, possono comunque avere la loro utilità. A questo scopo, ferma restando l'ampia variabilità che caratterizza i soggetti idealmente presi come riferimento per la formulazione dei modelli teorici, vi sono delle condizioni patologiche e fisiologiche la cui conoscenza è importante per individuare i loro limiti di applicazione.
- fattori fisiologici nel neonato: l'elevata percentuale di acqua corporea (75-80%) e la bassa percentuale di tessuto adiposo possono influenzare la distribuzione di certi farmaci. Inoltre, la quantità e la capacità legante dell'albumina neonatale sono minori rispetto all'adulto e la quota libera di alcuni farmaci, quali penicilline, fenitoina (barbiturico), fenobarbitale, può essere aumentata predisponendo ad effetti indesiderati.
- fattori patologici: il legame dei farmaci con le proteine plasmatiche può modificarsi in diverse condizioni patologiche. Negli stadi avanzati di malattie epatiche croniche, come la cirrosi, si può determinare una condizione di ipoalbuminemia con diminuzione del legame proteico dei
 farmaci acidi che si legano preferenzialmente a questa proteina. L'aumento della quota libera di alcuni farmaci, tra cui fenitoina e salicilati, può comportare, almeno teoricamente, un aumentato rischio di effetti indesiderati in epatopazienti ipoalbuminemici. Un'analoga situazione si può presentare nell'insufficienza renale in cui, per la diminuzione della concentrazione di albumina, per l'alterata struttura dei siti di legame e per l'accumulo di sostanze endogene che competono con i siti di legame proteico, si può verificare una significativa riduzione del legame di molti farmaci acidi, tra cui i salicilati, i sulfamidici, la fenitoina e la furosemide. Un'aumentata incidenza di effetti tossici sono stati riportati in pazienti ipoalbuminemici trattati con fenitoina.
farmaci acidi che si legano preferenzialmente a questa proteina. L'aumento della quota libera di alcuni farmaci, tra cui fenitoina e salicilati, può comportare, almeno teoricamente, un aumentato rischio di effetti indesiderati in epatopazienti ipoalbuminemici. Un'analoga situazione si può presentare nell'insufficienza renale in cui, per la diminuzione della concentrazione di albumina, per l'alterata struttura dei siti di legame e per l'accumulo di sostanze endogene che competono con i siti di legame proteico, si può verificare una significativa riduzione del legame di molti farmaci acidi, tra cui i salicilati, i sulfamidici, la fenitoina e la furosemide. Un'aumentata incidenza di effetti tossici sono stati riportati in pazienti ipoalbuminemici trattati con fenitoina.
- fattori fisiologici nell'anziano: la distribuzione dei farmaci è soggetta a modificazioni. L'acqua corporea totale diminuisce, mentre la percentuale di tessuto adiposo aumenta.
 Inoltre, si presentano notevoli variazioni nel volume di distribuzione; ciò è dovuto al minor potere legante delle proteine plasmatiche, alle modificazioni del metabolismo epatico, alla ridotta inducibilità delle attività enzimatiche, alla rallentata eliminazione renale. E' anche da considerare che la maggior sensibilità dei recettori e la minore efficienza dei meccanismi omeostatici espongono più facilmente il paziente anziano al rischio di effetti collaterali, soprattutto di tipo neurotossico. A queste problematiche, si aggiungono possibuli reazioni idiosincrasiche che diventano più frequenti. Nel paragrafo seguente queste problematiche sono discussa più approfonditamente.
Inoltre, si presentano notevoli variazioni nel volume di distribuzione; ciò è dovuto al minor potere legante delle proteine plasmatiche, alle modificazioni del metabolismo epatico, alla ridotta inducibilità delle attività enzimatiche, alla rallentata eliminazione renale. E' anche da considerare che la maggior sensibilità dei recettori e la minore efficienza dei meccanismi omeostatici espongono più facilmente il paziente anziano al rischio di effetti collaterali, soprattutto di tipo neurotossico. A queste problematiche, si aggiungono possibuli reazioni idiosincrasiche che diventano più frequenti. Nel paragrafo seguente queste problematiche sono discussa più approfonditamente.
le modificazioni della farmacocinetica nel paziente anziano
Circa l'86% della popolazione anziana è affetta da una patologia cronica ed il 50% di questa presenta l'associazione di due o più quadri patologici. Così, è naturale che l'età geriatrica sia di per sé caratterizzata dall'assunzione costante di numerosi farmaci. Fra questi devono essere ricordati: i cardiovascolari, gli analgesici, i sedativi ed i tranquillanti. Tutti questi farmaci presentano effetti collaterali e interazioni farmacologiche ed il soggetto anziano appare più esposto alla comparsa di reazioni avverse, la cui incidenza aumenta con il numero di farmaci assunti.
Nella pratica medica potrebbe risultare di particolare interesse poter stabilire degli orientamenti relativi al trattamento farmacologico, soprattutto per quanto riguarda i pazienti anziani. D'altra parte, per questa tipologia di pazienti, anche a motivo della variabilità delle loro condizioni di salute, non è possibile proporre delle linee guida standard.
Tuttavia, vi sono alcuni criteri che il medico dovrà tener presente e che sono legati alle variazioni fisiologiche della farmacocinetica indotte dall'età, quali soprattutto le modificazioni dei compartimenti corporei e del conseguente volume di distribuzione dei farmaci, cui si associa una riduzione della funzionalità degli organi destinati al loro metabolismo ed escrezione.
Tutto questo fa sì che il paziente anziano possa essere particolarmente sensibile ai farmaci di più comune impiego e possa manifestare con maggior facilità reazioni avverse derivanti dal loro uso.
In particolare, nel paziente anziano si osserva una diversità di risposta al trattamento farmacologico a causa delle modificazioni della composizione corporea proprie di quest'età. Il processo di invecchiamento altera, infatti, la farmacocinetica prevalentemente attraverso le variazioni qualitative e quantitative dei tessuti corporei. Intorno ai 60 anni il peso corporeo aumenta approssimativamente del 25% nell'uomo e del 18% nella donna, per diminuire però negli anni successivi fino ai livelli dell'età giovanile o ancora inferiori. A questa variazione ponderale fa riscontro anche una variazione della composizione corporea: nei soggetti ultraottantenni si assiste in media ad una perdita di circa 6 kg di massa muscolare, contro un incremento di circa 5 kg di massa lipidica; la quota idrica subisce una riduzione di circa il 12%. Queste modificazioni sono più evidenti nel sesso femminile.
Il volume plasmatico si riduce con l'età in modo proporzionale alla riduzione del volume idrico e l'entità di questo evento può risultare particolarmente importante negli individui debilitati. Le alterazioni della composizione corporea modificano la distribuzione
dei farmaci: la diminuzione della massa magra e della quota idrica, associata all'aumento della massa totale lipidica, fa sì che nei pazienti anziani i farmaci idrosolubili presentano un minor volume di distribuzione (aumenta la concentrazione plasmatica), mentre quelli liposolubili presentano un maggior volume di distribuzione.
Il volume di distribuzione dei farmaci risente anche delle modificazioni del loro legame con le proteine. Nell'invecchiamento si verifica una diminuzione del 20% della concentrazione plasmatica di albumina, probabilmente a causa della ridotta produzione epatica di questa proteina. Questo determina un incremento della quota libera dei farmaci che si legano alle proteine e dunque un aumento del volume di distribuzione. Vi possono essere, inoltre, anche delle modificazioni qualitative nella capacità di legarsi dell'albumina, in grado di determinare un aumento delle concentrazioni delle sostanze farmacologicamente attive e, pertanto, nel soggetto anziano si può assistere a risposte esagerate ai farmaci che normalmente
si legano alle proteine.
Con l'età, quindi, la diminuzione del legame proteico può aumentare gli effetti tossici o farmacologici: ad esempio farmaci come i salicilati, la digossina, la furosemide, l'indometacina, la penicillina, il dicumarolo, che hanno un elevato legame proteico, nel soggetto anziano possono presentare un incremento dei loro effetti. In generale, a queste osservazioni, si può aggiungere una distinzione legata alle caratteristiche chimiche del farmaco.
- farmaci idrosolubili: la somministrazione di un farmaco ad un soggetto anziano può dar luogo a concentrazioni farmacologiche più elevate di quelle che ci si aspetterebbe. Questo fenomeno potrebbe dare l'impressione di un'accresciuta sensibilità al farmaco. Pertanto, nell'impiego di alcuni farmaci idrosolubili, come ad esempio il paracetamolo ed altri antiinfiammatori non steroidei, ampiamente utilizzati come analgesici, si dovranno utilizzare dosaggi inferiori.
- farmaci liposolubili: le modificazioni della composizione corporea alterano quella frazione della massa corporea che agisce come "reservoir" per le sostanze liposolubili. Così se il volume di distribuzione di un farmaco liposolubile è significativamente aumentato, il tempo necessario per la sua eliminazione, anche qualora la clearance plasmatica sia immodificata, risulta prolungato. Se poi fosse alterata anche la clearance, per una diminuzione della massa epatica o per una diminuzione della perfusione renale, l'effetto finale si tradurrebbe in una maggiore emivita ed in un aumento della durata degli effetti clinici del farmaco. Questa variabilità nel paziente anziano, rende opportuno iniziare con la somministrazione di piccole dosi di farmaco e adattare i dosaggi in base alle risposte individuali.
somministrazione di farmaci in età pediatrica
La fisiologia pediatrica comporta delle differenze nei processi di metabolismo ed eliminazione; di queste differenze occorre tener conto adattando i dosaggi dei farmaci somministrati.
esempio: un medico ha prescritto un farmaco la cui dose media per adulti è 250 mg al giorno. Per un bambino alto 100 cm e 25 kg di peso, il dosaggio di questo farmaco dovrebbe essere ridotto.
Come primo passo, si deve determinare la body surface area (BSA):
kg0.425 · cm0.725 · 0.007184 = BSA
con i dati proposti, risulta: BSA = 250.425·1000.725· 0.007184 = 0.795
(La formula proposta è uno dei metodi per il calcolo dei BSA; i risultati ottenuti con altre formule possono variare)
successivamente, si applica la formula* :
dose adulto · (BSA/1.73) = dose bambino approssimativa
nel caso in esame, risulta: 250 · (0.795/1.73) = 114.88 mg
*Gerald, M. C., & OBannon, F. V.
(1988). Nursing pharmacology and therapeutics. (2nd ed.). Englewood Cliffs, NJ:
Prentice Hall Incorporated.

Il calcolo sviluppato può essere eseguito utilizzando il form sottostante che ha unicamente finalità didattiche e non dovrebbe essere usato per applicazioni mediche (per calcolare BSA si usano generalmente nomogrammi). Si consideri che non tutti i farmaci sicuri per gli adulti possono essere adattati alla diversa fisiologia del bambino; ciò vale particolarmente per i neonati.
interazioni fra farmaci
Poiché il legame dei farmaci con le proteine plasmatiche è in genere debole e non selettivo, molti farmaci con proprietà chimico-fisiche simili possono competere tra loro e con sostanze endogene per gli stessi siti di legame. Farmaci con elevata affinità per i siti di legame possono spiazzare farmaci con minore affinità aumentando la quota di farmaco attivo a livello dei siti d'azione.
In realtà, il significato clinico dei fenomeni di spiazzamento è stato ridimensionato e le implicazioni tossicologiche sono rare e comunque legate ad un indice terapeutico modesto. Infatti, l'aumento della quota libera può rinforzare l'effetto farmacologico, ma tale potenziamento è solo transitorio in quanto aumenta anche la quota di farmaco disponibile per il metabolismo e l'escrezione. Nella maggior parte dei casi il fenomeno è quindi di modesta entità. Però, possono manifestarsi conseguenze cliniche quando il farmaco spiazzato sia legato per più del 90% , abbia un piccolo volume di distribuzione ed un basso indice terapeutico, soprattutto poi se lo spiazzamento è associato ad un fenomeno di inibizione metabolica. In queste condizioni l'aumento della quota libera può portare alla comparsa di effetti tossici (cfr. cinetica non lineare).
monitoraggio della terapia farmacologica
Individuati i limiti di applicazione dei modelli farmacocinetici, si tratta di stabilire quando sia necessaria e utile - al di fuori della sperimentazione chimica - la loro utilizzazione per controllare ed ottimizzare il trattamento terapeutico.
Un criterio fondamentale per decidere se monitorizzare la concentrazione ematica dei farmaci richiede che vi sia una correlazione fra concentrazioni ematiche ed effetti terapeutici e/o tossici. La maggior parte dei farmaci agisce con meccanismo recettoriale e quindi gli effetti riscontrati sono proporzionali al numero di interazioni con il sito d'azione e dunque alla concentrazione di farmaco nei tessuti. Ora, poiché non è possibile determinare la concentrazione di farmaco nel sito di azione, la quantità presente nel corpo viene stimata misurando la concentrazione in uno dei liquidi biologici di perfusione: plasma, sangue, siero. Questa pratica trova il suo fondamento sul fatto che il plasma rappresenta il compartimento centrale che riceve il farmaco dal sito di somministrazione durante il processo di assorbimento e lo cede a tutti gli organi e tessuti compresi quelli deputati all'eliminazione. Quindi, se esiste una correlazione fra concentrazione tissutale di farmaco ed effetto terapeutico, questa sarà anche riferibile al tasso ematico e ciò permetterà di ottenere informazioni utili anche dal compartimento ematico.
Il monitoraggio dei livelli ematici dei farmaci è indicato in alcuni casi1:
- insuccesso terapeutico a dispetto di posologie ritenute adeguate;
- durante affezioni epatiche o renali;
- trattamento con più farmaci;
- dosi elevate di farmaco per via endovenosa;
- prematurità o dismaturità;
- terapie superiori a dieci giorni in caso di trattamento con antibiotici aminoglicosidici (attivi contro le infezioni sostenute da batteri Gram negativi);
- edemi importanti;
- gravidanza e obesità.
Tuttavia, nel caso di degenza in regime non ospedaliero, l'utilità del monitoraggio è limitata dalla non immediata accessibilità dei risultati delle analisi di laboratorio; pertanto, si dovrebbe consigliare il ricovero quando sia documentata l'utilità del monitoraggio, e questo è riconducibile alla presenza di tre condizioni specifiche:
- ridotto indice terapeutico;
- elevata variabilità dei livelli ematici per una determinata dose;
- correlazione fra l'entità della concentrazione ematica e gli effetti terapeutici e/o tossici.
Il controllo posologico per i farmaci che verificano le citate condizioni diventa perentorio in alcune situazioni cliniche:
- patologie che possono comportare notevoli variazioni dei parametri farmacocinetici da un giorno all'altro;
- pazienti con insufficienza renale, epatica o cardiocircolatoria;
- soggetti obesi o in stato di gravidanza;
- i neonati.
1Richens S.A. and Warrington S., When should plasma levels drug be monitored? Drugs 1979, 17, 488-500
tossicocinetica negli avvelenamenti *
Lo studio della farmacocinetica fornisce un criterio di valutazione razionale, specie per quanto concerne le procedure intese a modificare l'assorbimento, la distribuzione o la eliminazione dei tossici. Purtroppo, studi di farmacocinetica applicata alla tossicologia clinica sono stati finora condotti solo per un limitato numero di sostanze. Per quanto concerne i farmaci, si fa spesso riferimento ai parametri cinetici misurati in volontari o pazienti dopo somministrazione di dosi terapeutiche, quindi si estrapolano i dati ad ipotetiche situazioni di sovradosaggio. E' ovvio, però, che questo criterio può condurre a conclusioni erronee e richiede in ogni caso molta cautela.
In generale, il profilo farmacocinetico nel sovradosaggio può differire da quello tipico delle dosi terapeutiche. Ciò a volte dipende da fenomeni di saturazione dei sistemi preposti al metabolismo o alla escrezione. In altri casi, la cinetica è modificata da fattori quali l'acidosi, l'ipotermia, il danno epatico o renale, l'insufficienza respiratoria o cardiocircolatoria, che spesso sono presenti quali intrinseche espressioni di tossicità della sostanza.
- assorbimento: poiché una quota cospicua degli avvelenamenti avviene per ingestione e in questo caso l'assorbimento gastro-intestinale dei tossici riveste particolare importanza. A volte, la cinetica di assorbimento presenta caratteristiche del tutto anomale dopo ingestione di dosi elevate: ad esempio, negli avvelenamenti da farmaci anticolinergici (antidepressivi triciclici, antiparkinsoniani, ecc.) l'attività propulsiva nel tratto gastro-enterico può essere fortemente depressa, cosicché il tempo di transito e il contatto del farmaco con la superficie assorbente dell'intestino vengono prolungati.
Varie categorie di sostanze tra cui beta-bloccanti, antidepressivi triciclici, fenotiazine e il metadone, vanno incontro ad un intenso metabolismo pre-sistemico ("first-pass" intestinale e/o epatico) i cui meccanismi vengono saturati ad alte dosi. Dopo ingestione di quantità elevate
dei suddetti farmaci, la frazione della dose che giunge in circolo può superare di molto quella attesa in base alla cinetica tipica del dosaggio terapeutico.
L'uso del carbone attivato (v. Tab. II), l'irrigazione intestinale e il trattamento con purganti, es. sodio solfato, sorbitolo, citrato di magnesio (Shannon et al.,1986), sono talora raccomandati negli avvelenamenti per ingestione allo scopo di limitare l'accesso dei tossici nel compartimento
sistemico. Esiste altresì evidenza che il trattamento ripetuto con carbone blocca il circolo entero-epatico o entero-enterale di certe sostanze e aumenta quindi la loro clearance sistemica. Questo vale, ad esempio, per la digossina, il fenobarbitale, la teofillina, la ciclosporina, il methotrexate, il fenilbutazone, la carbamazepina e il diazepam.
- distribuzione: in presenza di alte concentrazioni plasmatiche di un tossico, il legame che quest'ultimo contrae con le proteine può essere saturato con conseguente aumento della frazione che circola libera nel plasma. Da ciò derivano importanti implicazioni tossicologiche poiché il volume di distribuzione, la velocità di eliminazione e l'accesso del tossico nei siti recettoriali vengono modificati.
I tessuti poco perfusi (muscoli, tessuto adiposo) fungono spesso da deposito delle sostanze lipofile, le quali vengono concentrate a tale livello per essere poi cedute lentamente nel distretto extracellulare. Il lento equilibrio tra i compartimenti intra- ed extracellulare è all'origine dell'effetto "reservoir" che talora si osserva quando l'eliminazione terapeutica dei tossici viene accelerata con procedure drastiche, quali la emodialisi o la emoperfusione: il trattamento comporta la progressiva diminuzione dei livelli ematici della sostanza i quali, tuttavia, tornano ad innalzarsi allorché il trattamento viene interrotto. Tale situazione è stata spesso riscontrata nella terapia emodialitica dell'intossicazione da sali di litio (Jacobsen et al. 1987).
metabolismo: il metabolismo dei composti chimici esogeni ha conseguenze che dipendono dalla attività dei metaboliti: ha significato detossicante se i metaboliti sono inattivi o meno attivi della sostanza di origine.
Si assiste invece alla comparsa o alla accentuazione della tossicità quando il composto di origine, di per sé poco attivo o inattivo, subisce un processo di attivazione metabolica.
La tossicità per attivazione ha due aspetti caratteristici:
- la sintomatologia si manifesta con una certa latenza che riflette il tempo necessario perché i metaboliti tossici vengano generati fino a raggiungere concentrazioni critiche nei tessuti bersaglio;
- mancano di solito correlazioni tra i livelli ematici del farmaco e lo stato del paziente, le cui condizioni possono anzi aggravarsi man mano che diminuiscono i livelli ematici della sostanza assorbita. Il deterioramento funzionale degli organi preposti al metabolismo ha influenze sulla cinetica dei tossici in quanto prolunga la loro permanenza nell'organismo. Nel corso degli avvelenamenti, ha particolare importanza la depressione dell'attività farmaco-metabolizzante del fegato che può essere causata sia da lesioni parenchimali indotte dal tossico sia da squilibri emodinamici che riducono la perfusione epatica.
L'attivazione farmacologica dei processi di detossicazione epatica è stata oggetto di numerosi studi in prospettiva terapeutica. Il corredo degli enzimi farmaco-metabolizzanti può essere, in effetti, aumentato mediante somministrazione di farmaci induttori, quali il fenobarbitale (è un induttore enzimatico, pertanto l'efficacia di alcuni farmaci - anticoagulanti, steroidi surrenali, antibiotici, contraccettivi orali e anticonvulsivanti come la fenitoina - può essere ridotta per accelerazione progressiva del metabolismo). Tale procedura, tuttavia, oltre ad essere non priva di effetti secondari, richiede somministrazioni ripetute nell'arco di alcuni giorni, cioè tempi troppo lunghi per essere compatibile con l'intervento d'urgenza nella intossicazione acuta. Esistono induttori meno tossici, quali i flavoni, che agiscono con rapidità in quanto attivano il corredo enzimatico pre-esistente. Non risulta, tuttavia, che tali sostanze siano state oggetto di studio in relazione a possibili impieghi in tossicologia clinica. La procedura corrente per impedire la generazione di metaboliti tossici è comunque di tipo farmacologico.
- eliminazione: l'eliminazione ha un ruolo centrale nella tossicocinetica clinica. La terapia di molti avvelenamenti è principalmente diretta a sostenere le funzioni vitali e a proteggere la funzionalità degli organi emuntori onde consentire la eliminazione fisiologica del tossico. Nelle intossicazioni più gravi trovano talora impiego, compatibilmente con le caratteristiche cinetiche del veleno, procedure specifiche intese ad accelerare la rimozione della sostanza o dei suoi metaboliti dall'organismo.
Il calcolo della emivita plasmatica rende talora possibile la stima approssimativa dei tempi necessari perché i livelli ematici della sostanza scendano nel paziente a valori sub-tossici. Tuttavia, molte sostanze presentano ad alte dosi cinetiche di eliminazione non lineari a causa della saturazione del metabolismo e/o della escrezione (Tabella I). In questi casi, il calcolo della emivita non ha alcun valore pratico se non si dispone di dati certi sulla dose totale assorbita.
Le biomembrane sono più permeabili alle sostanze non ionizzate che a quelle ionizzate. Pertanto, per le sostanze eliminate dal rene, il riassorbimento nei tubuli diminuisce quanto più il composto si presenta ionizzato nell'urina tubulare. Dato che il grado di ionizzazione degli acidi deboli aumenta se questi si trovano in ambiente alcalino e quello delle sostanze basiche aumenta nelle soluzioni acide, è possibile incrementare l'escrezione renale dei tossici acidi o basici attraverso procedure che determinano opportune variazioni del pH e del flusso urinario.
L'ingestione di tossici in quantità potenzialmente letali, la presenza di sintomi gravi, il riscontro di livelli ematici straordinariamente elevati, l'evidenza che i processi di eliminazione sono saturati, la comparsa di deficit delle vie escretrici, il progressivo deterioramento dello stato del paziente nonostante la terapia intensiva sono tutte condizioni che, in linea di principio, suggeriscono il ricorso alle misure drastiche di eliminazione terapeutica, quali l'emodialisi e l'emoperfusione. Tuttavia, è spesso difficile prevedere quali pazienti potranno trarre effettivi benefici di queste procedure anche in relazione al loro intrinseco potenziale di morbilità.
In ogni caso, il ricorso alla depurazione extracorporea è applicabile solo per le sostanze che hanno caratteristiche farmacocinetiche compatibili. Per i tossici con spiccata tendenza alla diffusione nei tessuti, forte affinità per le proteine plasmatiche e ampio volume di distribuzione, è poco probabile che l'aumento della clearance determinato dalla emodialisi o dalla emoperfusione si associ a risultati clinicamente significativi, data la modesta frazione della dose che si trova in forma libera nel circolo.
avvelenamenti da formulazioni farmaceutiche a lento rilascio *
L'impiego in medicina di preparati a lento rilascio è sempre più diffuso. L'ingestione di dosi tossiche comporta particolari problemi date le peculiari caratteristiche cinetiche dei princìpi attivi di queste formulazioni. L'assorbimento è di solito più lento e prolungato ed il picco di concentrazione da accumulo viene raggiunto non prima di 24-36 ore. La tossicità si manifesta dopo un periodo di latenza più o meno lungo e le manifestazioni regrediscono piuttosto lentamente al termine della fase acuta a causa dell'assorbimento protratto della sostanza nel lume intestinale.
Nella terapia dell'avvelenamento hanno importanza la somministrazione ripetuta di carbone e l'irrigazione intestinale, da praticarsi possibilmente già alla comparsa dei primi sintomi, al fine di limitare la quantità di principio attivo che passa in circolo.
Da quanto discusso, è ovvio che l'analisi tossicocinetica costituisce una guida per affrontare con criteri scientifici i problemi diagnostici e terapeutici della tossicologia clinica. Il laboratorio chimico-tossicologico, avvalendosi di apparecchiature e tecniche analitiche sempre più sensibili, precise, selettive e veloci, grazie alla tossicocinetica può fornire indicazioni di quegli aspetti quantitativi e temporali degli avvelenamenti la cui conoscenza è importante per un intervento clinico razionale.
Purtroppo, nella pratica corrente, raramente l'analisi chimico-tossicologica viene effettuata attraverso indagini che comprendano, ove necessario, l'identificazione e il dosaggio dei metaboliti attivi. D'altra parte, solo per pochi composti si conoscono le caratteristiche farmacocinetiche in situazioni di sovradosaggio. Anche per i farmaci più noti, i dati della letteratura, per la variabilità delle condizioni sono spesso di difficile interpretazione. Per esempio, la dose o il momento della esposizione al tossico sono molte volte imprecisati; in altri casi, i prelievi per le analisi tossicologiche risultano essere stati effettuati per tempi troppo brevi o, ancóra, manca l'adeguata valutazione di patologie pre-esistenti o di altri fattori capaci di modificare la farmacocinetica.
Tabella I. Esempi di sostanze che
presentano cinetiche di saturazione
( Rif.: Mullen, 1980; Rosenberg et al., 1981.)
| Salicilici |
Prednisolone |
| Paracetamolo |
Diossano |
| Teofillina |
Alcol etilico |
| Fenitoina |
Acido triclorofenossiacetico (2,4,5 T) |
| Chinidina |
Cloruro di vinile |
| Amilobarbitone |
Cloralio idrato |
| Sulfametazina |
Tricloroetanolo |
Tabella II. Adsorbimento dei tossici
da parte del carbone attivato "in vitro"
( modificato da Neuvonen e Olkkola, 1988.)
| elevato |
moderato |
scarso |
| Amfetamine |
Aspirina |
Cianuri |
| Antidepressivi |
Benzene |
Etanolo |
| Antiepilettici |
Bifenili policlorurati |
Ferro |
| Antistaminici |
Cherosene |
Glicole etilenico |
| Atropina |
Clorpropamide |
Litio |
| Barbiturici |
Diclorometano |
Metanolo |
| Benzodiazepine |
DDT |
Sostanze caustiche |
| Chinidina |
Disopiramide |
|
| Chinina |
Fenolo |
|
| Cimetidina |
Malathion |
|
| Fenotiazine |
Mexiletina |
|
| Fenilbutazone |
Paracetamolo |
|
| Fenilpropanolamina |
Tolazamide |
|
| Furosemide |
Tolbutamide |
|
| Glibenclamide |
Glipzide |
|
| Glucosidi digitalici |
Indometacina |
|
| Oppiacei |
|
|
| Piroxicam |
|
|
| Stricnina |
|
|
| Teofillina |
|
|
tratto e modificato da http://anestit.unipa.it/esiait/1098_01.htm
cronofarmacologia
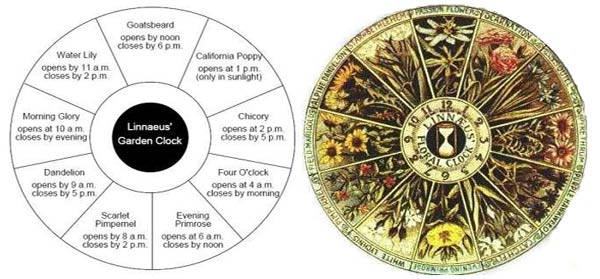
Carl Linnaeus (1707-1778) con lunghi anni di osservazioni, scoprì che molte piante si schiudono e si aprono periodicamente ed i loro tempi variano da specie a specie. Disponendo i fiori lungo un ideale sequenza giornaliera, Linnaeus realizzò un grazioso segnatempo, un "orologio floreale" che descrisse nellla sua opera Philosophiu botunicu (Vienna, 175 1): 274-275.
Ovviamente Linnaeus ignorava la risposta delle piante alla differente lunghezza del giorno (fotoperiodo), ma per la particolare latitudine di Uppsala (60" N), molte delle piante che aveva selezionato erano adattate a brevi notti e fotoperiodi di 12 h o più. Il suo elenco comprende anche specie di tipo intermedio, che fioriscono indipendentemente dalla durata della luce diurna. Queste specie foto-indipendenti (per es. Taraxacum officinale) non sono adatti come segnatempo giacché il loro tempo di apertura varia con le stagioni.
Questa apertura e chiusura periodica dei fiori è legata all'interazione di un ritmo endogeno e la lunghezza del giorno (segnale luminosità/buio) ed accade che le piante siano capaci di misurare il tempo a partire dallo stimo0lo luminoso. Tuttavia, non abbiamo alcuna conoscenza dei recettori coinvolti che sono racchiusi nei fitocromi. Il processo di apertura e chiusura riguarda certamente cambiamenti nel turgore di piccoli gruppi di cellule; questi cambiamenti possono essere influenzati dalla temperatura e dall'umidità.
Durante la prima metà del 19 .mo secolo, i giardinieri provarono a costruire orologi floreali, ma con scarso successo dal momento che molte delle piante elencate da Linnaeus non fioriscono nella stessa stagione.
A questo punto, è interessante - ai fini della tecnologia farmaceutica - un seppur limitato cenno alle conoscenze sui ritmi circadiani, che cercano una qualche correlazione fra effetto di un farmaco e ora della sua somministrazione.
La somministrazione di corticosteroidi, per esempio, può essere resa più razionale se si tiene conto del fatto che le surrenali presentano il max di attività verso le prime ore del mattino.
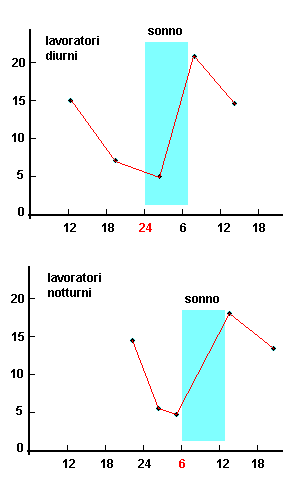 Nei due diagrammi a destra, sono mostrate le concentrazioni plasmatiche di corticosteroidi riferite a soggetti che lavorano durante il giorno ed a lavoratori notturni.
Nei due diagrammi a destra, sono mostrate le concentrazioni plasmatiche di corticosteroidi riferite a soggetti che lavorano durante il giorno ed a lavoratori notturni.
Fondandosi su queste osservazioni, sono state sviluppati due medicinali:
- la pillola trifasica (contraccettivo orale) caratterizzata da un contenuto ormonale particolarmente basso e da una composizione diversificata in tre fasi.
- Synachten Depot (Tetracosactide: ACTH sintetico), che agisce sulla corteccia surrenale, ha la funzione di provocare la secrezione di cortisone endogeno (che ha notoriamente potere antinfiammatorio, e può contribuire a ridurre, oppure eliminare, la necessità di trattamenti farmacologici o strumentali esogeni diretti allo stesso scopo) con un ritmo più naturale di quanto ottenibile ricorrendo ad una somministrazione cortisonica. Ovviamente, nei casi in cui necessiti un effetto più rapido, il trattamento cortisonico è preferibile.
Da quanto accennato, è chiaro che si sta diffondendo la cronofarmacologia, la farmacologia connessa cioè non soltanto con le dosi dei farmaci da somministrare, ma anche con il momento in cui i farmaci vanno somministrati. Si è ritenuto infatti per secoli che somministrando identiche dosi di una medicina, l'efficacia di essa fosse costante in tutti; a parte i casi di sensibilità, differenziata da persona a persona, verso la medicina stessa (per cui un identico farmaco in certi casi fà bene, in altri fà poco e in altri ancora dà disturbi) è risultato che uguali dosi a uguali intervalli di tempo danno tuttavia effetti diversi, mentre effetti costanti si possono ottenere con dosi diverse somministrate in tempi diversi. Per esempio, la levotiroxina per l'ipotiroidismo deve essere assunta al mattino; al contrario, gli antinfiammatori vanno assunti la sera, in modo da migliorare le condizioni al risveglio. L'interferone è più sicuro se somministrato di sera in quanto al mattino darebbe origine ad una sindrome parainfluenzale.
Questa relatività di effetti, più che al farmaco, è dovuta all'organismo che lo riceve e al momento in cui lo riceve; al mattino, per essere espliciti, un farmaco può dimostrarsi più attivo che alla sera perché, al mattino, le funzioni dell'organismo si trovano in una fase di attività che rende l'organismo stesso più ricettivo alla sua efficacia. pertanto, la conclusione dei farmacologi clinici, è che la tempo-dipendenza dell'azione dei farmaci è non soltanto quantitativa ma anche qualitativa; in altre parole, la somministrazione dei farmaci andrà sempre riferita in futuro ai "tempi del corpo" e non al "tempo dell'ambiente", quello segnato dall'orologio.
D'altra parte, se è possibile, grazie ai dovuti accertamenti di cui si è appena parlato, modificare le terapie secondo le caratteristiche individuali dei pazienti, si sta pure dimostrando che sarà possibile manipolare i ritmi biologici del nostro corpo, in modo da ottenere una maggiore disponibilità dell'organismo nei confronti di determinate cure.
 Moltissime persone sperimentano il jet lag. Per esempio, se si va in Australia non è possibile modificare immediatamente l'orologio biologico per adattarlo al diverso fuso orario in quanto la capacità di regolazione del nostro bioritmo arriva al massimo ad unora al giorno. Così, nel caso vi sia una differenza di otto ore, non potendo modificare più di una sola ora al giorno, sarà necessaria almeno una settimana per adattarsi al nuovo orario: fino ad allora non si riescirà a dormire bene, perché l'orologio biologico è ancóra sincronizzato sull'orario italiano.
Moltissime persone sperimentano il jet lag. Per esempio, se si va in Australia non è possibile modificare immediatamente l'orologio biologico per adattarlo al diverso fuso orario in quanto la capacità di regolazione del nostro bioritmo arriva al massimo ad unora al giorno. Così, nel caso vi sia una differenza di otto ore, non potendo modificare più di una sola ora al giorno, sarà necessaria almeno una settimana per adattarsi al nuovo orario: fino ad allora non si riescirà a dormire bene, perché l'orologio biologico è ancóra sincronizzato sull'orario italiano.
 1 1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
| HOME PAGE |
Marcello Guidotti, copyright 2010
questa pagina può essere riprodotta su qualsiasi supporto o rivista purché sia citata la fonte e l'indirizzo di questo sito (ai sensi degli artt. 2575 e 2576 cc. Legislazione sul diritto d'autore). Le fotografie sono tratte da siti web e sono, o possono ritenersi, di pubblico dominio purché utilizzate senza fini di lucro. Le immagini di prodotti presenti nel sito hanno unicamente valenza esemplificativa oltre che, eventualmente, illustrare messaggi fuorvianti e non vi è alcun richiamo diretto o indiretto alla loro qualità e/o efficacia il cui controllo è affidato alle autorità regolamentatorie.