 |
 |
|
Una delle prime ipotesi, che associava la struttura chimica di una molecola alla sua funzione, fu formulata da Blake nel 1848. Blake propose che l'attività biologica di alcuni sali metallici fosse dovuta alla loro componente metallica, piuttosto che all'intero complesso (ad esempio, il piombo nell'acetato di piombo è tossico mentre l'acetato di sodio in miscela con l'acido acetico può essere aggiunto ai cibi in qualità di conservante, E262). Questo concetto ricevette il sostegno di Arrhenius nel 1884, quando introdusse la sua teoria della dissociazione elettrolitica.
La relazione fra struttura molecolare ed azione farmacologica fu riconosciuta con evidenza da Brown e Fraser nel corso del tardo diciannovesimo secolo. Essi dimostrarono che la quaternizzazione (l'aggiunta di un quarto gruppo alchilico) di una serie di alcaloidi si traduceva in una loro inversione di effetto, da muscolo contrattore a muscolo rilassante; così, inferirono l'esistenza di un rapporto tra l'azione fisiologica di una sostanza e la sua struttura chimica.
Alle soglie del ventesimo secolo, Meyer e Overton, con un loro lavoro sull'importanza della lipido solubilità nell'azione dei farmaci, contribuirono anche alla futura teoria recettoriale. La loro teoria, che costituiva un tentativo di spiegare l'attività delle diverse strutture chimiche che esistono all'interno delle classi generali di farmaci ipnotici e anestetici, correlava gli effetti farmacologici con una proprietà fisica (ad esempio, la solubilità dei lipidi), piuttosto che con la relazione struttura-attività. Oggi conosciamo l'erroneità di questa teoria (per esempio non spiega perché due enantiomeri dello stesso ipnotico, come il tiopentale, pur avendo la stessa solubilità lipidica differiscano notevolmente nella loro efficacia) e puntiamo l'attenzione alla struttura chimica, che è un fattore determinante delle proprietà fisiche.
Il concetto di farmaci che agiscono sui recettori è generalmente accreditato agli studi di John Langley nel 1878. Langley, mentre studiava gli effetti antagonisti dell'atropina contro la salivazione indotta dalla pilocarpina, scrisse: "c'è qualche sostanza o alcune sostanze nelle terminazioni nervose o nelle ghiandole cellulari in grado di formare composti con atropina e con pilocarpine". Successivamente fece riferimento a questo fattore/i come una "sostanza ricettiva". Nonostante questa osservazione, la parola "retettore" è stato introdotto nella letteratura medica, solo alla fine del secolo da Paul Ehrlich.
Ehrlich fondò la sua ipotesi sulla sua esperienza nel trattamento delle malattie infettive con farmaci derivati dall'industria tedesca dei coloranti. Egli postulò che un farmaco potesse avere un effetto terapeutico solo se dotato della "giusta sorta di affinità". In particolare, scrisse specificamente "che la combinazione di gruppi di molecole proptoplasmiche a cui si ancora la molecola attiva sarà d'ora in poi indicata come recettore". Ehrlich immaginò i recettori come facenti parte di catene laterali in cellule di mammifero, oggi sappiamo che i siti di legame con il farmaco possono essere parte di ogni componente cellulare.
La sperimentazione di una serie di farmaci indicavano chiaramente che le molecole farmacologicamente attive, al fine di produrre i loro effetti, si concentravano su piccole aree specifiche di cellule. Questi effetti evidenziano:
|
|
|
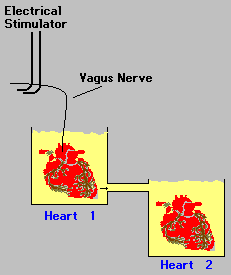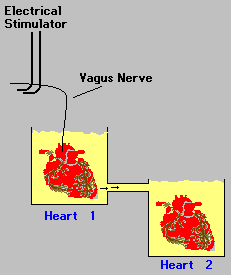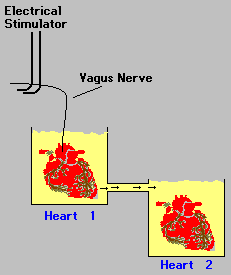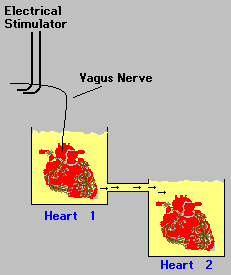
Loewi stimolò il nervo vago del primo cuore per produrvi un rallentamento del battito e potè osservare che anche il secondo cuore dopo un certo ritardo rallentava. Loewi formulò l'ipotesi che la stimolazione elettrica che aveva rallentato il primo cuore rilasciasse nella soluzione una sostanza chimica che fluendo nel secondo recipiente rallentava anche il secondo cuore [il ritardo nella comparsa dell'effetto esclude una propagazione di tipo elettrico - NdT]. Egli chiamò questa sostanza "vagusstoff". Oggi sappiamo che questa sostanza chimica è un neurotrasmettitore chiamato acetilcolina. Il lavoro pioneristico di Loewi segnò la nascita della neurofarmacologia: la scienza che studia come i farmaci interagiscono con il sistema nervoso.
*tratto da: 1997 da Mark E. McCourt - www.psych.ndsu.nodak.edu/mccourt/Psy486/Biochemistry
Posta questa premessa, il passo successivo implica la conoscenza della tipologia e localizzazione dei recettori.
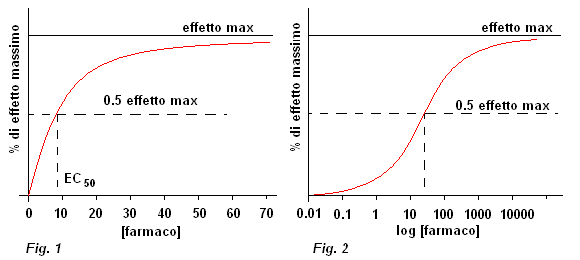
Nel diagramma a sinistra (Fig. 1), la relazione tra concentrazione di farmaco e risposta, un'iperbole rettangolare, è data da un modello matematico che abbiamo già incontrato nel trasporto attivo: l'equazione di Michaelis Menten.
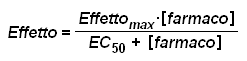 (1)
(1)
dove: Effettomax è la massima risposta del sistema al farmaco;
EC50 è la concentrazione di farmaco che produce una risposta pari al 50 per cento dell'effetto massimo.
Nella formulazione proposta dall'equazione, a seconda che EC50 sia molto maggiore o minore di [farmaco] si ottiene l'andamento in Fig. 1. Tuttavia, in questo caso, occorre individuare la relazione da attribuire al confronto fra i termini a denominatore: EC50 e [farmaco] .
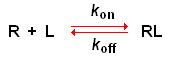
la costante di equilibrio, fornisce:
la frazione di recettore totale, Rtotale = [RL]+[R], che esiste come complesso RL è:
sostituendo [RL] ricavata dalla (2) nella (3), si ottiene il risultato cercato:
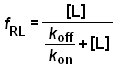
quando è raggiunta la metà dell'effetto massimo, EC50, la frazione di farmaco impegnata nel legame recettoriale deve essere: fRL = 0.5 (i recettori sono occupati al 50%) risulta [L] = [farmaco] = koff / kon come riportato sul grafico di Fig. 3
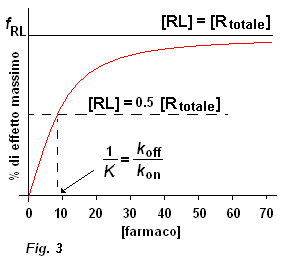
poiché l'intensità della risposta di un recettore ad un farmaco è proporzionale alla concentrazione di farmaco legato, si può affermare che:
La frazione di recettori occupata è di rilevante interesse in quanto è da questa che dipende l'effetto di un farmaco. Così, se noi misuriamo l'effetto massimo (dove circa il 100% dei recettori sono occupati) e conosciamo la frazione impegnata allora possiamo determinare l'effetto per ogni data concentrazione di farmaco. Questo effetto sarà dato proprio dall'eq. 1 (generalizzata per un ligando, L, e con EC50 = K ):
L'affinità di un ligando, L, per il recettore è tanto maggiore quanto minore è K, cioé quanto più lentamente il complesso farmaco recettore si dissocia.
In generale l'effetto degli antagonisti farmacologici deriva dall'impedire all'agonista di legarsi al recettore ed attivarlo. Gli antagonisti possono essere competitivi (spiazzati reversibilmente dagli agonisti) o non competitivi (spiazzati non reversibilmente dagli agonisti). Con un modello rozzo ma intuitivo, il comportamento di un agonista nei confronti di un recettore viene comunemente paragonato ad un sistema chiave-serratura. Questa rappresentazione deriva dal fatto che i legami di Van der Waals ed i legami idrogeno sono molto deboli e quindi le associazioni molecolari dipendenti da questi legami e dotate di una certa stabilità e specificità, devono implicare un elevato numero di legami per unità di superficie di interazio fra molecole. Per questa ragione le macromolecole mostrano cavità e protuberanze che si compenetrano una nell'altra in un modo analogo a quello della chiave e della serratura.
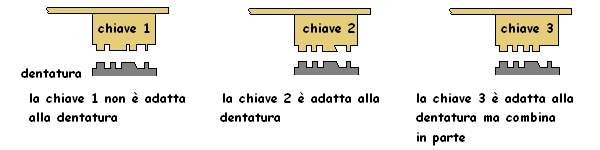
Nel primo caso (chiave 1), è rappresentata la condizione di non interazione fra agonista (o antagonista) e recettore: la chiave (agonista o antagonista) non gira la dentatura della serratura (recettore).
Nel secondo caso (chiave 2), è rappresentata la condizione di interazione fra agonista (o antagonista) e recettore: la chiave (agonista o antagonista) si adatta alla serratura e quindi gira la dentatura (recettore).
Nel terzo caso (chiave 3), è rappresentata la condizione di interazione parziale fra agonista (o antagonista) e recettore: la chiave (agonista o antagonista) si adatta parzialmente alla serratura e non gira la dentatura completamente.
L'eq. 4 non permette di discriminare fra i vari tipi di antagonisti, per questo si introduce il parametro "attività intrinseca", α:
La relazione di Michaelis-Menten applicata al trasporto attivo, ha la stessa formulazione matematica della relazione applicata alla combinazione farmaco recettore. Tuttavia, il significato è profondamente differente, e questo può essere chiarito con le due immagini che seguono: una evocativa del trasporto attivo; l'altra, della competizione agonista-antagonista.
 |
 |
|
| trasporto attivo: il carrier non può trasportare più elementi del previsto, e alla saturazione ogni elemento aggiunto comporta l'uscita di un altro elemento. | attivazione recettoriale: la sostituzione di un componente con un altro, quando riesce, comporta risultati diversi. In questo caso la sostituzione di un metro cubo di calcestruzzo con uno di legname comporta una maggiore emissione di CO2 | .
Un ligando è un antagonista non competitivo (o insormontabile) quando si lega irreversibilmente al recettore di modo che i suoi effetti non possono essere completamente annullati aumentando la concentrazione di agonista e quindi, al contrario dell'inibizione competitiva, la risposta massima all'agonista non può essere ristabilita. Questo significa che l'affinità dell'antagonista per il recettore può essere talmente elevata da impedire l'accesso all'agonista.
Da un punto di vista pratico, la durata di azione di un antagonista non competitivo dipende più dalla capacità dell'organismo di produrre nuovi recettori che dalla velocità con cui viene eliminata la quota di antagonista non legato ai recettori.
Un antagonista competitivo, C, compete con l'agonista, A, per il legame con il recettore, R, secondo lo schema seguente:
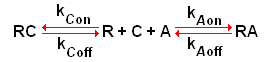
Per la legge di azione di massa, aumentando la concentrazione di C, l'equilibrio si sposterà a sinistra riducendo la quantità dell'efficace complesso recettore-agonista, AR. Al contrario, aumentando la concentrazione di agonista, l'equilibrio si sposterà a destra riducendo la quantità dell'improduttivo complesso recettore-antagonista.
Poiché un antagonista competitivo, C, non produce effetti, per misurarne il potere inibente verso l'agonista, si definisce il rapporto di dose, Rd :
 |
se la concentrazione di agonista in presenza di antagonista è la stessa che in sua assenza, allora l'antagonista non è efficace ed Rd = 1 ; però è evidente che tanto maggiore è Rd, tanto più rilevante è l'effetto dell'antagonista e maggiore è la concentrazione di agonista necessaria per ottenere un dato effetto. Si può dimostrare che Rd è legato alla concentrazione di antagonista, C, tramite la relazione di Schild, dove KC è la costante di dissociazione dell'antagonista.
Riprendiamo in esame l'eq. 1 :
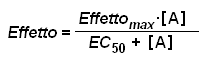 (1)
(1)come abbiamo discusso, la (1) descrive la relazione tra la concentrazione di un singolo ligando e il suo grado di combinazione con il recettore. Quando è presente, con concentrazione C, un secondo ligando antagonista che compete reversibilmente con il primo per lo stesso recettore, vale la relazione (ottenuta combinando le eq. 1 e 6) seguente:
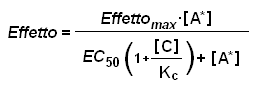 (7)
(7)si noti che la EC50 è ridotta di un fattore legato alla concentrazione di antagonista tramite il rapporto di dose dato dalla (6); per ottenere lo stesso effetto che si avrebbe in assenza di antagonista, è necessario che la concentrazione [A] di agonista aumenti (eq. 7), fino al valore [A*]= Rd [A]
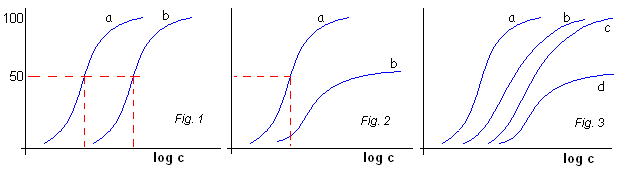
grafico in Fig. 1 : la curva a è ottenuta con concentrazioni crescenti di agonista; la curva b è ottenuta con concentrazioni crescenti dello stesso agonista in presenza di una concentrazione fissa di antagonista. L'antagonismo è di tipo competitivo;
grafico in Fig. 2 : la curva a è ottenuta con concentrazioni crescenti di agonista; la curva b è ottenuta con concentrazioni crescenti dello stesso agonista in presenza di una concentrazione fissa di antagonista non competitivo che legandosi irreversibilmente al recettore impedisce di raggiungere l'effetto massimo;
grafico in Fig. 3 : la curva a è ottenuta con concentrazioni crescenti di agonista; le curve b e c sono ottenute con concentrazioni crescenti dello stesso agonista in presenza di una concentrazione fissa di antagonista non competitivo che si lega irreversibilmente al recettore. Sebbene i recettori siano diminuiti, le curve b e c mostrano che l'effetto massimo è ancora raggiungibile. Solamente quando un numero sempre crescente di recettori è occupato irreversibilmente, l'effetto massimo, curva d non è più raggiungibile.
In presenza di un antagonista non competitivo, l'affinità dell'agonista per il recettore non varia, però l'effetto massimo non è più raggiungibile a causa della diminuita disponibilità dei recettori che non possono più essere liberati per competizione.
L'antagonismo è di tipo misto quando la risposta all'agonista in presenza di basse concentrazioni di agonista insormontabile, che sottrae irreversibilmente recettori al farmaco agonista, può raggiungere comunque il massimo. Una tale evidenza sperimentale è in aperto contrasto con la teoria dell'occupazione recettoriale (secondo la quale l'effetto massimo raggiungibile dall'agonista è conseguente all'occupazione di tutti i recettori presenti); tuttavia, può trovare una spiegazione ammettendo l'esistenza di recettori di riserva. In pratica, esistono organi o tessuti che apparentemente contengono un numero di recettori superiori a quello necessario a un agonista dotato di attività intrinseca massima (a = 1) per evocare una risposta ottimale. La presenza di questi recettori potenzia così anche la risposta dell'organo bersaglio all'agonista parziale ( a < 1) in quanto il maggior numero di recettori occupati tende a compensare la minore attività intrinseca.
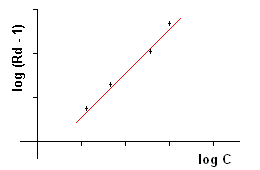 riportando in scala logaritmica il primo membro in ordinate; log [C] in ascisse, si ottiene una retta dove l'intercetta sulle ordinate è data da - log KC.
riportando in scala logaritmica il primo membro in ordinate; log [C] in ascisse, si ottiene una retta dove l'intercetta sulle ordinate è data da - log KC.Quando il coefficiente angolare della retta di Schild è 1, l'antagonismo è competitivo e reversibile. Questo implica che l'agonista sta agendo su un singolo tipo di recettore e che il tessuto non ha meccanismi di assorbimento (uptake) per l'agonista, che potrebbero falsare le osservazioni. Si può anche concludere che l'antagonista determina un parallelo spostamento a destra della curva di risposta dell'agonista senza diminuzione della risposta massima.
Quando il coefficiente angolare della retta di Schild non è 1, allora vi sono tre possibilità:
| 4 | |||||||
Marcello Guidotti, copyright 2005-2009-2011
questa pagina può essere riprodotta su qualsiasi supporto o rivista purché sia citata la fonte e l'indirizzo di questo sito (ai sensi degli artt. 2575 e 2576 cc. Legislazione sul diritto d'autore). Le fotografie sono tratte da siti web e sono, o possono ritenersi, di pubblico dominio purché utilizzate senza fini di lucro. Le immagini di prodotti presenti nel sito hanno unicamente valenza esemplificativa oltre che, eventualmente, illustrare messaggi fuorvianti e non vi è alcun richiamo diretto o indiretto alla loro qualità e/o efficacia il cui controllo è affidato alle autorità regolamentatorie.