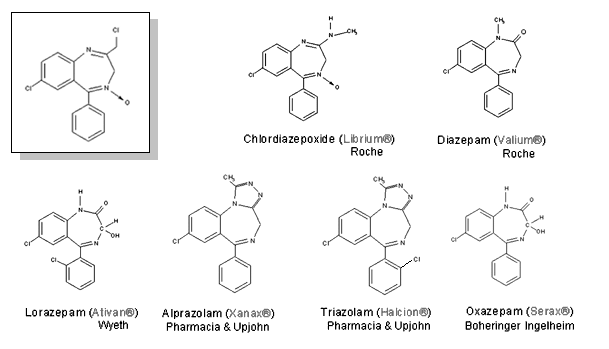
alcune benzodiazepine coperte con brevetto di selezione da aziende differenti.
La Legge sulle invenzioni e la Convenzione sulla concessione di brevetti europei (CBE) prevedono che tutti i brevetti che hanno per oggetto un composto chimico, devono possedere requisiti di novità, originalità ed industrialità. Le industrie farmaceutiche ricorrono a vari tipi di brevetto:
|
Soprattutto in campo farmaceutico, a meno delle restrizioni imposte dall'esistenza di un brevetto di prodotto, esistono in commercio farmaci molto simili per caratteristiche ed effetti. Un esempio classico è offerto delle benzodiazepine: sostanze cui appartiene la stragrande maggioranza degli ansiolitici e dei sonniferi. La prima benzodiazepina sintetizzate è il clordiazepossido (basato sulla modifica di una struttura nota e non coperta da brevetto, il cui scheletro è racchiuso nel riquadro): il clordiazepossido è stato brevettato, ma non è stata brevettata la futura famiglia delle benzodiazepine; infatti, sostituendo un atomo o un gruppo di atomi, alla molecola di partenza (non brevettata) si sono ottenute le altre benzodiazepine, che sono state brevettate da aziende diverse (v. figura sotto).
 Si noti che alprazolam e triazolam sono brevetti Pharmacia & Upjoh : l'alprazolam è la modifica di una molecola capostipite (nel riquadro) non coperta da brevetto; tuttavia, il suo significativo aumento di complessità la rende praticamente un nuovo prodotto, sicché le modifiche a questa molecola (addizione di Cl in posizione 2'), che ha portato al triazolam, possono essere realizzate solo dal titolare del brevetto. Al contrario, le molecole di oxazepam (Boehringer Ingelheim) e lorazepam (Wyeth) differiscono per la presenza in quest'ultima di un cloro in posizione 6'. Queste osservazioni mostrano quanto sia delicata la questione dei brevetti (con gli inevitabili contenziosi legali) richiesti per prodotti che, se ottenuti da comuni reazioni chimiche sulla molecola capostipite (nel riquadro), possono dare spazio a molecole "me too". Il diagramma a sinistra riporta le ingiunzioni avanzate da aziende farmaceutiche dell'UE al fine di contrastare l'immissione di medicinali da parte di aziende concorrenti.
Si noti che alprazolam e triazolam sono brevetti Pharmacia & Upjoh : l'alprazolam è la modifica di una molecola capostipite (nel riquadro) non coperta da brevetto; tuttavia, il suo significativo aumento di complessità la rende praticamente un nuovo prodotto, sicché le modifiche a questa molecola (addizione di Cl in posizione 2'), che ha portato al triazolam, possono essere realizzate solo dal titolare del brevetto. Al contrario, le molecole di oxazepam (Boehringer Ingelheim) e lorazepam (Wyeth) differiscono per la presenza in quest'ultima di un cloro in posizione 6'. Queste osservazioni mostrano quanto sia delicata la questione dei brevetti (con gli inevitabili contenziosi legali) richiesti per prodotti che, se ottenuti da comuni reazioni chimiche sulla molecola capostipite (nel riquadro), possono dare spazio a molecole "me too". Il diagramma a sinistra riporta le ingiunzioni avanzate da aziende farmaceutiche dell'UE al fine di contrastare l'immissione di medicinali da parte di aziende concorrenti.
Tecnicamente, col "senno del poi", le aziende avrebbero potuto richiedere un brevetto di sbarramento (protegge una famiglia di composti caratterizzati dallo stesso gruppo funzionale di base, al quale, presumibilmente, potrebbero corrispondere effetti terapeutici simili). Questo brevetto è utile nel caso di importanti innovazioni riguardanti la scoperta di una nuova famiglia di molecole: la sola presenza di un brevetto di selezione permetterebbe ad altre aziende, infatti, di produrre molecole analoghe, aggirando e/o eliminando il vantaggio competitivo che dovrebbe essere garantito all'azienda innovatrice.
 Il brevetto di sbarramento può sembrare eccessivo; tuttavia, è perfettamente legale se la formula di struttura proposta è omogenea, cioè si riferisce ad una sola classe di sostanze; se ognuno dei possibili sostituenti è rappresentato con esempi (cosa abbastanza difficile) ed il loro numero è definito; se ognuno dei prodotti è effettivamente preparabile. Il brevetto di sbarramento può sembrare eccessivo; tuttavia, è perfettamente legale se la formula di struttura proposta è omogenea, cioè si riferisce ad una sola classe di sostanze; se ognuno dei possibili sostituenti è rappresentato con esempi (cosa abbastanza difficile) ed il loro numero è definito; se ognuno dei prodotti è effettivamente preparabile.
|
D'altra parte, l'oggetto del brevetto deve essere attuato: se questa condizione necessaria (olre ad altre che la rendano sufficiente) non è rispettata, ogni interessato che ne faccia richiesta, può ottenere una licenza obbligatoria a suo favore.
La mancanza del brevetto di sbarramento, ha condotto sia alla nascita di altre molecole più efficienti di quelle originarie ma anche di prodotti di cui non si sentiva la mancanza perché senza reali vantaggi rispetto ai precedenti (in inglese si chiamano me-too products, prodotti "ci sono anch'io").
|
I brevetti discussi, fatta eccezione per il brevetto di prodotto, si riferiscono a molecole per le quali è scaduta la protezione brevettuale. Diversamente, l'azienda che, per esempio, volesse registrare l'associazione di un farmaco protetto da brevetto con un altro a brevetto scaduto per la quale esperienze cliniche occasionali - non frutto di specifica sperimentazione (in quanto interdetta dal brevetto di prodotto) - hanno dimostrato un'azione sinergica, deve accordarsi con l'azienda detentrice del brevetto di prodotto per ottenerne la concessione d'uso.
|
Di rilevante importanza è il certificato di protezione supplementare dei farmaci preso in considerazione dall'accordo TRIPS (Trade-Related Aspects of Intellectual Property Rights) che ha stabilito gli standard di protezione della proprietà intellettuale che porterà, in quei Paesi che hanno sottoscritto l'accordo, alla protezione per i brevetti chimici e farmaceutici.
In tale accordo si fa riferimento alla necessità di armonizzare la durata della protezione brevettuale che attualmente è di venti anni dal deposito della domanda. D'altra parte, per i farmaci tale periodo è di 8-10 anni. Questo perché per commercializzare il prodotto è necessaria l'Autorizzazione all'Immissione in Commercio (AIC) da parte del Ministero della Salute, dopo aver presentato la documentazione inerente i risultati delle sperimentazioni chimiche sul farmaco.
|
La tutela brevettuale garantisce che l'azienda detentrice del brevetto possa commercializzarlo in esclusiva per almeno 20 anni. Tenendo conto che occorrono almeno 10-12 anni perché un nuovo medicinale arrivi sul banco della Farmacia, questo significa che all'azienda rimangono solo 8 per ripagarne (complessivamente) 20. Per questa ragione, è stato istituito il "certificato di protezione supplementare" (v. avanti)
|
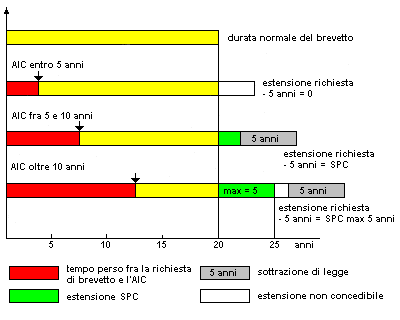 La figura schematizza il funzionamento del SPC: il meccanismo non comporta alcun vantaggio qualora l'AIC avvenga entro 5 anni dalla richiesta di brevetto, mentre si ottiene il periodo di copertura brevettuale maggiore (25 anni) in caso di autorizzazione concessa dopo 10 anni. |
Il certificato di protezione supplementare è il titolo in forza del quale si prolunga la durata dell'esclusiva brevettuale limitatamente al prodotto (v. contenzioso MIT) medicinale o fitosanitario ottenuto dal brevetto, al fine di far recuperare il tempo intercorso tra la data della domanda di brevetto e l’autorizzazione all’immissione in commercio del prodotto.
L'SPC ha una validità, decorrente dal termine di scadenza del brevetto, pari al periodo compreso tra la data della domanda di brevetto e l’autorizzazione all’immissione in commercio del prodotto detratti 5 anni e, comunque, non può avere durata superiore a 5 anni.
Tra i fattori principali che in Italia hanno sfavorito lo sviluppo del mercato dei medicinali equivalenti vi è sicuramente la eccessiva lunghezza della copertura brevettuale che ha, nel nostro paese, una durata di 20 anni escluso il suo prolungamento. Infatti la brevettabilità dei prodotti farmaceutici, si è avuta in Italia a partire dal 1978 e successivamente con l'emanazione del DPR 338/1979 prima e con la legge 349/91 poi, è stato istituito il Certificato di Protezione Complementare (CPC) abrogato dal Regolamento CEE 1768/92 e sostituito con il Supplementary Protection Certificate (SPC). La differenza tra queste due normative risiede nella durata massima dell'estensione concessa alla fine della durata legale del brevetto che, per il CCP è non superiore a 18 anni (ridotto progressivamente in base a successivi provvedimenti), mentre per il SPC non può superare i 5 anni.
Infatti, a norma della direttiva 2001/83/CE (Art. 10), La Domanda semplificata di AIC per i medicinali generici [richiede]
1. In deroga all'articolo 8, comma 3, lettera l), e fatta salva la disciplina della tutela della proprietà industriale e commerciale, il richiedente non e' tenuto a fornire i risultati delle prove precliniche e delle sperimentazioni cliniche se può dimostrare che il medicinale e' un medicinale generico di un medicinale di riferimento che e' autorizzato o e' stato autorizzato a norma dell'articolo 6 da almeno otto anni in Italia o nella Comunità europea.
2. Un medicinale generico autorizzato ai sensi del presente articolo non può essere immesso in commercio, finche' non sono trascorsi dieci anni dall'autorizzazione iniziale del medicinale di riferimento. Un chiaro riferimento a tale divieto e' contenuto nel provvedimento di AIC.
3. Se il medicinale di riferimento non e' stato autorizzato in Italia ma in un altro Stato membro della Comunità europea, il richiedente indica nella domanda il nome dello Stato membro in cui il medicinale di riferimento e' autorizzato o e' stato autorizzato. L'AIFA chiede all'autorità competente dell'altro Stato membro di trasmettere, entro un mese, la conferma che il medicinale di riferimento e' autorizzato o e' stato autorizzato, insieme alla composizione completa del medicinale di riferimento e, se necessario, ad altra documentazione pertinente, con riferimento, in particolare, alla data dell'AIC rilasciata nello Stato estero.
4. Il periodo di dieci anni di cui al comma 2 e' esteso ad un massimo di undici anni se durante i primi otto anni di tale decennio il titolare dell'AIC ottiene un'autorizzazione per una o più indicazioni terapeutiche nuove che, dalla valutazione scientifica preliminare all'autorizzazione, sono state ritenute tali da apportare un beneficio clinico rilevante rispetto alle terapie esistenti.
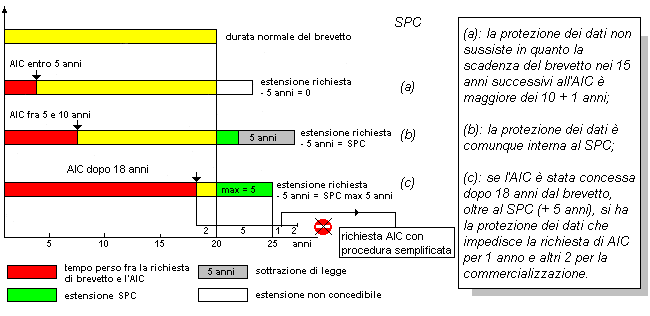
Questi commi prevedono, dunque, che la richiesta di AIC per un medicinale equivalente possa essere ottenuta senza fornire idonea documentazione clinica, ma unicamente tramite prove di bioequivalenza se l'AIC dell'originator è stata concessa almeno 8 anni prima. Comunque, per la commercializzazione del medicinale equivalente devono trascorrere 10 anni dalla AIC dell'originator (11 anni se ricorrono le condizioni del comma 4). Lo schema sottostante aiuta a chiarire le varie eventualità che si possono presentare. La situazione prevsita dall'Art. 10, si presenta per quei medicinali che hanno ottenuto l'AIC dopo 18 anni dalla concessione del brevetto; infatti, in questo caso, la protezione supplementare non può essere maggiore di 5 anni e quindi il brevetto scadrà dopo 7 anni. Dunque, il competitor potrà presentare domanda di AIC con procedura semplificata dopo un anno e potrà commercializzare il medicinale dopo altri 2 anni.
|
Occorre fare una precisazione fra quanto previsto in conseguenza del "caso Bolar" e la protezione dei dati: la possibilità di effettuare studi di bioequivalenza durante l'ultimo periodo di vita della protezione brevettuale, è differente dal percorso amministrativo richiesto per ottenere l'AIC. Nel caso (c) riassunto sopra, il competitor, sebbene nella disponibilità dei richiesti studi di bioequivalenza, non potrà presentare la domanda di AIC prima di quanto previsto dall'Art. 10. |
| 7 | ||||||
Marcello Guidotti, copyright 2003-2004-2005-2006-2010-2011
Le immagini di prodotti presenti nel sito hanno unicamente valenza esemplificativa oltre che, eventualmente, illustrare messaggi fuorvianti e non vi è alcun richiamo diretto o indiretto alla loro qualità e/o efficacia il cui controllo è affidato alle autorità regolamentatorie.